看文献时发现一个信息量很丰富的图片,于是研究了一下并进行复现,先放个成果图
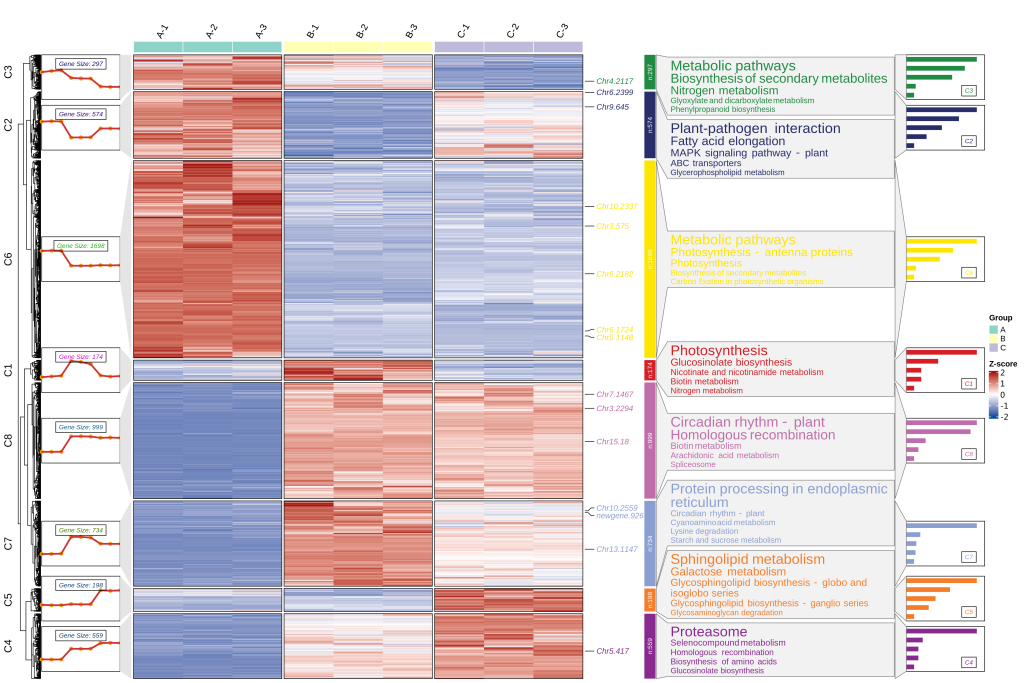
实现思路为,先对表达量数据做kmeans聚类分析进行分组(可使用其他分析如mfuzz, wgcna),然后对每组基因进行富集分析(KEGG,GO),然后使用ClusterGVis 包进行结果整合可视化。
ClusterGVis github地址: https://github.com/junjunlab/ClusterGVis
可以将包下下来,使用devtools::install_local('./path') 命令进行安装
PS : 安装ClusterGVis包时需要注意各依赖项的版本问题,我安装的是ClusterGVis 0.1.1版本的,其中使用了ComplexHeatmap的anno_block函数的align_to参数,低版本的ComplexHeatmap没有该参数,后面更新到了2.20.0版本的ComplexHeatmap才能用
# Argument importation
option <- function() {
disc <- "ClusterGVis"
parser <- ArgumentParser(description = disc)
parseradd_argument("--cluster",
type = "character",
required = T,
help = '输入kmeans结果文件,第一列是基因,第二列是clusterID,第三列往后是各样本表达量数据.')
parseradd_argument("-g", "--group",
type = "character",
required = T,
help = "输入样本分组文件,第一列是样本,第二列是分组.")
parseradd_argument('-o', '--outdir',
type = "character",
default = "./",
required = F,
help = '输出目录. [Optional, default: %(default)s]')
parseradd_argument('--focus',
type = "character",
required = F,
default = NULL,
help = '关注基因列表,一列数据,没有表头,可以不给,不给就不画关注基因. [Optional]')
parseradd_argument('--enrichRes',
type = "character",
required = F,
default = NULL,
help = '富集结果文件,可以不给,不给就不画富集结果,必须包含三列数据,表头为cluster,Description,P-value.')
parseradd_argument('--enrichTop',
type = "integer",
required = F,
default = 5,
help = '画富集结果时选用top个数画图,太多了会很挤,显示不全. [Optional, default: %(default)s]')
if (length(commandArgs(T)) == 0) {
parserprint_help()
quit(status = 1)
}
args <- parserparse_args()
return(args)
}
library(argparse)
# Get command line options
args <- option()
Col_L15 <- c("#8dd3c7","#ffffb3","#bebada","#fb8072","#80b1d3","#fdb462","#b3de69","#fccde5",
"#d9d9d9","#bc80bd","#ccebc5","#ffed6f","#928262","#0067A5","#4DBF4A","#fb9a99","#b2df8a")
# ComplexHeatmap 版本(2.4.3)低了用不了(anno_block 没有align_to参数)
library(ClusterGVis)
options(scipen = 100, stringsAsFactors = F)
rawdata <- read.table(argscluster, header = T, sep = '\t', quote = '', check.names = F)
#head(rawdata)
#ID cluster A-1 A-2 A-3 B-1 B-2 B-3 C-1 C-2 C-3
#Chr11.1255 1 0.0675 0.0328 0.1165 2.9284 2.9944 2.5721 0.1141 0.0412 0.0432
#Chr11.342 1 0.2846 0.083 0.2947 0.9262 1.1964 0.5811 0.1925 0.1739 0.2185
#Chr11.981 1 6.3054 8.2915 8.481 16.3796 16.2822 14.0238 4.2052 5.6431 3.8638
#Chr12.2476 1 1.4847 1.1787 0.3138 1.8628 0.3538 1.6086 0.7857 0.2592 0.504
colnames(rawdata)[1:2] <- c('gene', 'cluster')
rownames(rawdata) <- rawdatagene
sampleid <- read.table(argsgroup, header = T, sep = '\t')
#head(sampleid)
#Sample Group
#A-1 A
#A-2 A
#A-3 A
#B-1 B
#B-2 B
outdir <- argsoutdir
if (!dir.exists(outdir)) {
dir.create(outdir)
}
anadata <- rawdata[, c('gene', 'cluster', as.character(sampleid[, 1]))]
# zscore
anadata[, 3:ncol(anadata)] <- t(apply(anadata[, 3:ncol(anadata)], 1, function(x) (x - mean(x, na.rm = T)) / sd(x, na.rm = T)))
# 构造数据列表
pl <- list()
plwide.res <- anadata
# lone.res是长数据,用来单独画折线图,这里需要
#pllong.res <- 'none'
pltype <- 'kmeans'
plgeneMode <- 'none'
plgeneType <- 'none'
# 自己构造样本注释条,实现颜色自由,legend标题自由
colcolor <- Col_L15[1:length(unique(sampleid[, 2]))]
names(colcolor) <- unique(sampleid[, 2])
#sampleColor <- colcolor[sampleid[, 2]]
sampledf <- sampleid[, 'Group', drop = F]
rownames(sampledf) <- sampleid[, 1]
sampleanno <- ComplexHeatmap::HeatmapAnnotation(df = sampledf,
col = list(Group = colcolor),
gp = grid::gpar(col = 'white'),
show_legend = TRUE,
show_annotation_name = FALSE)
# 构建画图参数列表,通过列表来给主函数传参,想加参数自己添加列表元素就行了,不用每次都用主函数带一堆参数
paramsList <- list(object = pl,
plot.type = "both",
column_names_rot= 60,
show_row_dend = T,
annnoblock.text = T,
sample.group = sampleid[, 2],
ctAnno.col = jjAnno::useMyCol("stallion", n = length(unique(rawdatacluster))), # 使用包默认颜色画行注释
#sample.col = sampleColor,
HeatmapAnnotation = sampleanno,
show_parent_dend_line = F,
add.box = F,
add.point = T,
point.arg = c(19, 'orange', 'orange', 0.5),
add.line = T,
line.side = 'left',
#cluster.order = sort(unique(rawdatacluster)),
column_title =\'' # 不显示分组标题
)
# 获取感兴趣基因名
if (!is.null(argsfocus)) {
markgene <- read.table(argsfocus, header = F, sep = '\t')
paramsListmarkGenes <- as.character(markgene[, 1])
}
# 传富集结果和富集画图参数
if (!is.null(argsenrichRes)) {
enrichData <- read.table(argsenrichRes, header = T, sep = '\t', quote = '', check.names = F)
enrichDatacluster <- as.numeric(enrichDatacluster)
if (length(unique(enrichDatacluster)) != length(unique(rawdatacluster))) {
stop('每个cluster必须都有富集结果才能画富集图, 没有就不给enrichRes参数,只画热图')
}
enrichRes <- data.frame()
for (i in unique(sort(enrichDatacluster))) {
clusterEnrichdata <- enrichData[enrichDatacluster == i, ]
if (nrow(clusterEnrichdata) > 0) {
clusterEnrichdata <- clusterEnrichdata[order(clusterEnrichdata`P-value`), ]
clusterEnrichdata <- head(clusterEnrichdata, argsenrichTop)
enrichRes <- rbind(enrichRes, data.frame('id'= paste0('C', i), clusterEnrichdata[, c('Description', 'P-value')]))
}
}
colnames(enrichRes) <- c('id', 'term', 'pval')
# id列是C1,C1,C2,C2...
paramsListannoKegg.data <- enrichRes
paramsListadd.kegg.bar <- T
paramsListkegg.size <- 'pval'
paramsListby.kegg <- 'anno_link'
paramsListannoKegg.mside <- 'right'
paramsListtextbar.pos <- c(0.8, 0.2) # 将富集柱状图的cluster标签放到右下角,避免遮挡图片
clusterColor <- jjAnno::useMyCol("stallion", n = length(unique(enrichResid)))
names(clusterColor) <- unique(enrichResid)
paramsListkegg.col <- clusterColor[enrichResid] # 设置富集的颜色和cluster注释颜色保持一致
}
p <- do.call(visCluster, paramsList) # 使用do.call函数将参数列表传给画图主函数
cairo_pdf(paste0(outdir, '/clusterGVis.pdf'), width = 18, height = 12)
print(p)
dev.off()
png(paste0(outdir, '/clusterGVis.png'), width = 18, height = 12, units = 'in', type = 'cairo', res = 300)
print(p)
dev.off()
该代码是外置传参的,将代码复制成plot.R,可用于linux系统批量画图,使用示例:
Rscript plot.R --cluster kmeans_cluster.txt -g group.txt -o results --enrichRes KEGG.data.txt --focus focus.id